How Project Optimus has Reshaped Oncology Drug Development: Opportunities and Challenges
In the wake of the FDA’s Project Optimus, an initiative to reform the current approaches to dose selection and dose optimization for oncology drugs, the clinical development of cancer therapies will likely undergo a prolonged and costly transition from the conventional paradigm. While the new approaches are geared toward yielding better-tolerated drugs, these changes require additional resources and could lead to fewer drugs reaching the market.
As the FDA’s guidelines for drug development and accelerated approvals in oncology continue to evolve in favor of patient centricity, these changes pose certain challenges for drug developers and clinical trial sponsors. In 2021, the FDA launched Project Optimus1, signaling a strategic shift toward reforming the dose optimization and dose selection paradigm in oncology. This endeavor gained momentum through a collaborative FDA-ASCO workshop2 in May 2022, followed by an FDA draft guidance document3 in January 2023.
Project Optimus represents a significant attempt to revolutionize dose-finding and dose-optimization strategies early in the clinical development of oncology drugs. Before the launch of Project Optimus, dose-finding studies predominantly relied on the maximum tolerated dose (MTD) paradigm, which was originally designed for cytotoxic chemotherapy. However, this approach is grounded in linear dose response and focuses on severe dose-limiting toxicities (DLTs), often resulting in poorly tolerated drugs and adversely impacting patients’ quality of life. As the treatment landscape for patients with cancer has shifted toward diverse dose-response relationships through targeted and novel therapies, it has become imperative to recognize the limitations of the MTD approach. As a result of Project Optimus, the FDA emphasizes the necessity of conducting tailored dose-finding studies that explore a spectrum of doses to find a balance between a drug’s efficacy and safety and allow for a subsequent evaluation of the selected doses. This strategic shift aligns with the evolving nature of oncology therapeutics, promoting a more nuanced and informed approach to identifying optimal dosage regimens and resulting in safer, better-tolerated drugs overall.
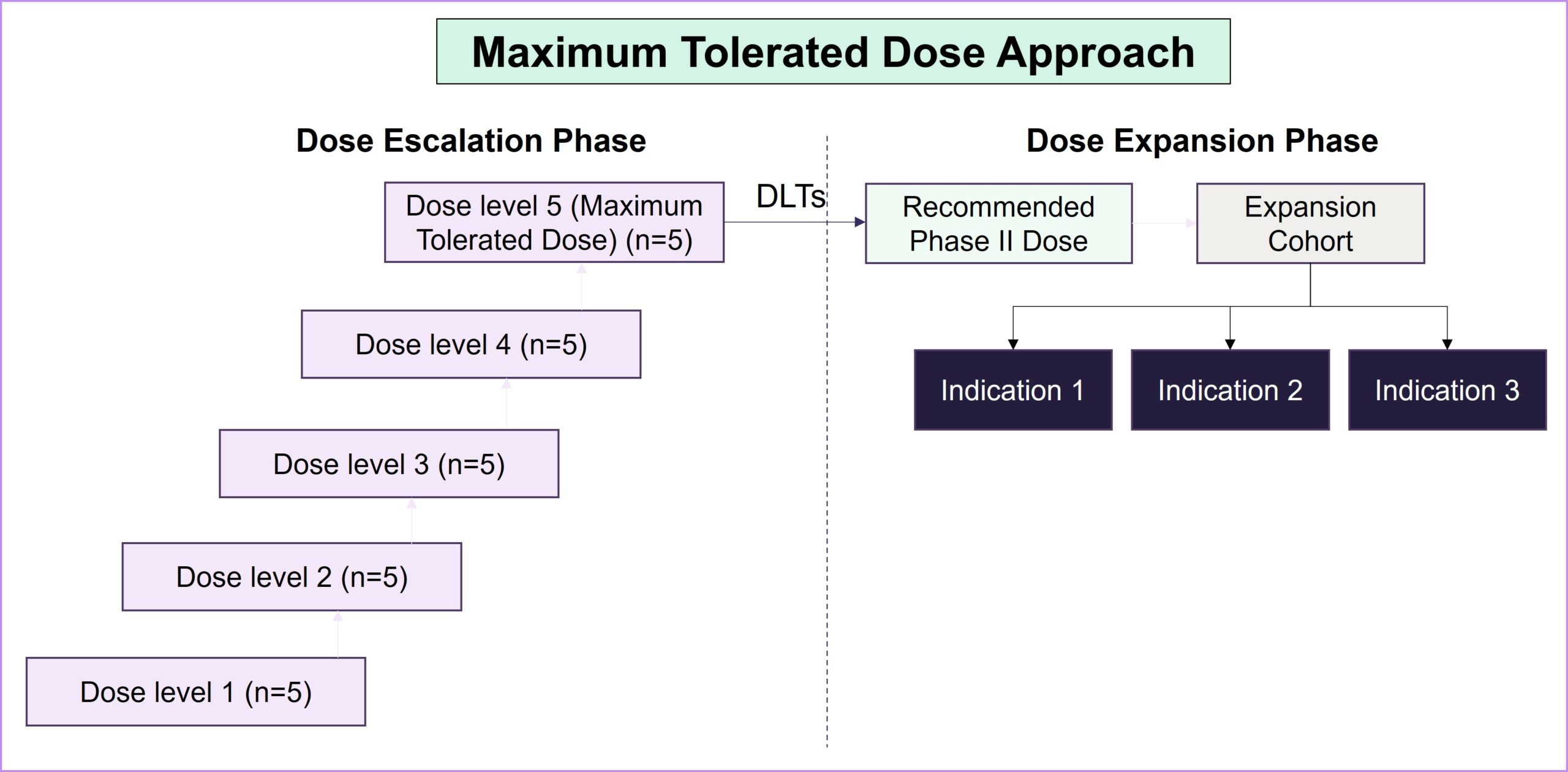
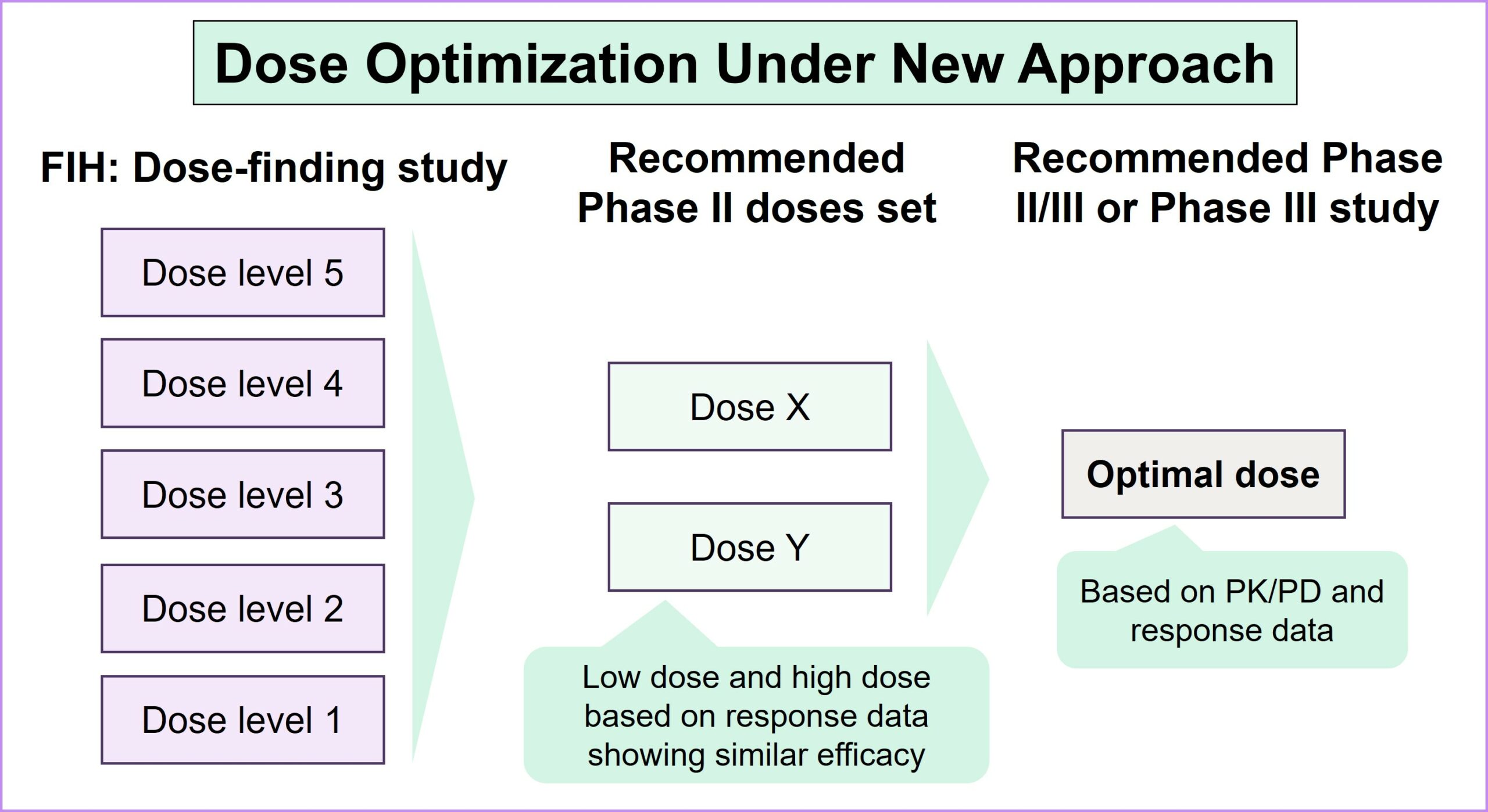
Strategic considerations in dose optimization for balancing efficacy, safety and patient outcomes: Key takeaways from the FDA’s guidance document
- Identification of dose optimization is a ‘must’, even in the fast lane of the FDA’s expedited programs: Expedited programs such as breakthrough designation will not be considered a valid rationale by the FDA to exclude dose optimization, providing sponsors with opportunities to discuss and plan their strategies more frequently and at different milestones. The FDA recommends continuous collaboration to synchronize dose-optimization strategies with a seamless development program for the accelerated clinical development of oncology products
- Dose selection for the clinical trial relies on both clinical and non-clinical data: This includes food effects early in drug development, pharmacokinetic sampling and analysis, and exposure-response analyses, along with anti-tumor activity, safety and tolerability. These factors will guide the selection for further evaluation, with clinical studies aiming to enroll a broader population for assessment across the relevant subpopulations
- A randomized, parallel-dose response study is recommended for comparing multiple doses: This ensures the evaluation of dose-response and exposure-response relationships, with a sufficient patient population for assessing the activity, safety and tolerability of each dose. Comparisons of multiple doses are recommended either prior to a pivotal study or as part of a registrational study. Regulators also recommend a parallel trial design in concept, with staggered lead-ins as required for safety
- Flexibility exists for dose selection in different scenarios; however, it is critical to generate supporting data:
- A safety protocol is required to define the measures to be taken in cases where there are frequent dose modifications or high numbers of SAEs
- Alternative dosage strategies should be considered to improve tolerability, including stepwise dosage
- Patient-reported outcomes (PROs) should be considered for evaluating tolerability in early-phase dose-finding studies
- PROs are typically assessed as secondary endpoints, and they do not drive successful outcomes. In a recent AP News story4, a doctor stated: “You get side effects, and then you have to stop the drug to recover from the side effects, and the tumor can grow.” A patient also explained: “The last thing we want to do is have our quality of life stolen from us.”
- A strong rationale is required when selecting a dose for evaluation in a pivotal study as different dosages could work differently across indications based on the tumor biology, patient segment and combination therapies used
Case studies under Project Optimus
- Amgen’s sotorasib (KRAS inhibitor): In May 2021, sotorasib was granted accelerated approval5 for patients with KRAS G12Cm NSCLC based on the response rate data from the dose-escalation portion of the CodeBreak100 study. As part of the accelerated approval, a post-marketing study was requested by the FDA to evaluate 240 mg QD (in two tablets) vs. 960 mg QD (in eight tablets) as similar pharmacokinetic/pharmacodynamic and efficacy data were seen across multiple dose levels in prior trials
- In Dec 20236, a post-marketing study for dose comparison was completed according to the FDA’s requirements, confirming the 960 mg QD dose of sotorasib in treating patients with KRAS G12Cm NSCLC under accelerated approval
- This example demonstrates the FDA’s redefined approach to new drug approvals, providing opportunities for sponsors to collaborate with the FDA in developing dose-optimization strategies for novel and targeted therapies
- GSK’s Blenrep: GSK’s Phase II DREAMM-147 study is another example of evaluating alternative dosage regimens, using Q4W and Q6W schedules to determine Blenrep’s risk-benefit profile in terms of lower corneal event rates with uncompromised efficacy in myeloma. However, the Phase II study was initiated only after multiple Phase III studies showed that ocular toxicity was a key concern, and futility in the confirmatory study resulted in the drug’s withdrawal from the market
- This underscores the importance of dedicating more time in initial studies to finding a tolerable dose as this is critical not only for patients’ quality of life but also for defining the success or failure of a drug
- Exelixis’ XB002 (TF-ADC): Exelixis8 is conducting a Phase I study to evaluate XB002 in solid tumors. In addition to the recommended dose of 2.25 mg/kg established in the dose-escalation phase, a lower dose of 1.7 mg/kg is being evaluated in the expansion phase to fulfill the FDA’s Project Optimus requirements for dose optimization
- Immunocore’s IMC-F106C (PRAME-A02): The company is collaborating with the FDA as part of Project Optimus to initiate a Phase III study in H1 20249. The first 90 patients will be randomized into three cohorts (two doses of IMC-F106C and a control arm). Based on the results from these cohorts, one dose will be selected for evaluation in a larger patient sample
- Immunomodulatory drugs such as IMC-F106C are increasingly common and are less likely to exhibit linear dose-response relationships
Implications for the industry: Improved drugs with a time and resource cost
- While this approach aims to support the robustness of clinical trials, it poses challenges, including delays and increased financial commitments
- Exploring multiple dosage levels to strike a balance between efficacy and safety holds the promise of delivering better drugs with reduced toxicity, leading to enhanced patient outcomes
- However, this could extend the drug development timeline given the increased patient enrollment requirement in early dose-finding studies and the need to manufacture additional dosage levels
- Additional investments, particularly for smaller companies, could lead to the discontinuation of programs that might have led to the development of effective drugs due to a lack of appetite for investment
- The inclusion of PROs as secondary outcomes could emerge as a pivotal component in clinical trials. This is emphasized in the FDA’s guidance document and underscores the significance of integrating PROs into tolerability assessments to gain a more comprehensive understanding of treatment outcomes
- The interpretation of toxicity studies in preclinical development will extend beyond conventional parameters, with an added emphasis on understanding the tolerability windows for drug metabolism and pharmacokinetics/drug absorption, distribution, metabolism and excretion (DMPK/ADME) studies
- In particular, the meticulous execution of minimum anticipated biological effect level (MABEL) studies will become increasingly important as the FDA underscores the requisite inclusion of preclinical data for dose selection
- With the gradual shift from MTD-based approaches to optimal dosage, significant changes will be seen in trial design methodologies
- Phase I studies will include emerging dosage levels, and Phase II studies will include parallel regimen testing. Phase II or Phase III studies will require larger patient populations than in the previous paradigm to generate the necessary registrational data. If the preferred dose was already identified in Phase I, this could lead to an accelerated approval at this stage
- Compared with previous paradigms where Phase I/II studies with dose escalation and dose expansion could sometimes directly pave the way for accelerated approval, this approach supports the initiation of a more robust study for confirmation and accelerated approval
More drugs vs. better drugs
To mitigate the adverse effects and poor quality of life associated with some current cancer therapies, regulatory bodies are intensifying their efforts to balance the safety and efficacy of oncology drugs, with the FDA going a step further in defining these therapies’ risk-benefit profiles. While the evolving parameters in drug development promise enhanced safety and responsiveness to patient needs, there is an inevitable trade-off: Fewer drugs will reach the market due to the need for increased investment and time. This presents a dilemma for pharmaceutical companies: They can choose to progress quickly with increased financial investment, or they take a more measured, stepwise approach that comes with the risk of falling behind their competitors.
References
- FDA’s Project Optimus
- FDA-ASCO workshop, May 2022
- FDA Guidance document, Jan 2023
- AP News, Feb 2024
- Sotorasib May 28, 2021, FDA approval
- Sotorasib regulatory update, Dec 2023
- DREAMM-14 ASCO publication, Jun 2022
- Exelixis Q2 2023 financial results, Aug 2023
- Immunocore Q2 2023 financial results, August 2023
Related Blogs


The Medical Affairs Leadership Catalyst: Unlocking Strategic Potential through Medical Planning
May 23, 2024